Targeting the Perfect Storm- Amino Acid Dysfunction in Post Exertional Malaise-
- Graham Exelby
- Dec 15, 2024
- 25 min read
Updated: Feb 13
Dr Graham Exelby. December 2024, revised February 2025
Introduction
Post-exertional malaise (PEM) represents a multifactorial challenge involving amino acid depletion, mitochondrial dysfunction, hypermetabolism, and brainstem hypoperfusion. These intertwined dysfunctions create a "perfect storm" that exacerbates symptoms in conditions such as POTS, chronic fatigue syndrome (CFS), long COVID, Gulf War Syndrome (GWS), and fibromyalgia (FMS). Investigations into PEM have provided promising avenues for understanding its complex pathophysiology and guiding targeted management strategies.
This document synthesizes evidence on PEM pathophysiology, focusing on aspartate’s critical metabolic role, glutamate imbalances, and the therapeutic implications of targeted interventions.
This is a preliminary paper reporting findings on ongoing research. It does not apply to all patients as all have different metabolic profiles. It does relate to the predominant changes we have found, and provides a basis for ongoing research into this very complex problem of PEM in Chronic Fatigue Syndrome, POTS, Long COVID and Fibromyalgia. It also carries implications for conditions such as Autism Spectrum Disorder and ADHD which will be explored as part of the ongoing research.
Pathophysiology of PEM Summary
Amino Acid Metabolism in PEM
Preliminary findings indicate persistent aspartate depletion post-exertion, aligning with the broader metabolic crisis hypothesis of PEM. Aspartate serves as a critical intermediary for:
TCA cycle anaplerosis, the metabolic process that replenishes the citric acid cycle’s intermediates which are essential for energy production
The malate-aspartate shuttle (MAS), critical for mitochondrial NADH transfer.
Nitrogen clearance in the urea cycle.
Dysregulated amino acid metabolism contributes to secondary glutamate imbalances. This dysfunction manifests as:
Impaired glutamate recycling and compartmentalization.
Neuroexcitation and excitotoxicity despite normal systemic glutamate levels.
Prolonged energy recovery due to slow replenishment of aspartate reserves.
Mitochondrial Dysfunction
PEM is characterized by reduced reserves of ATP, with prolonged recovery times due to impaired mitochondrial function. Evidence suggests a reliance on non-pyruvate-dependent TCA cycle intermediates, such as:
Branched-chain amino acids (e.g., leucine, isoleucine, valine).
Amino acids entering the TCA cycle as acetyl-CoA (e.g., lysine, phenylalanine, tryptophan).
Brainstem Hypoperfusion
Brainstem hypoperfusion has been implicated in autonomic dysfunction and PEM, as evidenced by SPECT imaging studies. Reduced perfusion impacts on global and regional cerebral blood flow regulation, as well as mitochondrial energy production within brainstem neurons
Findings from Wirth et al. (10) highlight a 24.5% reduction in brainstem blood flow upon positional changes in ME/CFS, correlating with impaired cognitive and autonomic regulation.
Nicotinamide and SIRT4 Modulation
Nicotinamide, a precursor for NAD⁺, modulates mitochondrial function via SIRT4 inhibition, relieving glutamate dehydrogenase (GDH) suppression and enhancing energy metabolism. Key effects include:
Restoring NAD⁺ levels, improving TCA cycle efficiency and amino acid metabolism.
Enhancing glutamate clearance, addressing excitotoxicity.
Supporting the malate-aspartate shuttle, mitigating aspartate depletion.
Clinical evidence supports liposomal nicotinamide riboside’s efficacy in reducing fatigue and stabilizing amino acid profiles in POTS and CFS.
Preliminary Study results
Preliminary results in amino acid testing after initial treatment with liposomal nicotinamide to modulate the SIRT4 signalling. Persistent aspartate depletion shown in follow up testing, points to a fundamental metabolic bottleneck in PEM, where aspartate is disproportionately consumed in an effort to maintain cellular energy homeostasis. This aligns with the broader metabolic crisis hypothesis of PEM, where a fragile energetic system is pushed beyond its threshold following exertion.
At the core of this dysfunction, aspartate’s depletion is driving an unresolved secondary glutamate imbalance, even when absolute glutamate levels appear normal. This suggests that the pathology is not simply a matter of excess glutamate production but rather a failure of glutamate utilization, recycling, and compartmentalization.
Figure 1. Comparison of Symptoms Severity among POTS, FMS, GWS and Long COVID
(25)(26)(27)(28)(29)(30)(31) CFS rating is 10/10 in fatigue and PEM.

Coat hanger pain reflects hypoxia in muscle groups in the neck and shoulders associated with mitochondrial dysfunction and muscle weakness. When muscles have inadequate oxygen there is a switch to anaerobic metabolism, with accumulation of lactic acid which causes the characteristic cramping and pain. The relationship between the two is shown with exercise, where in POTS, CFS and many Long COVID even minor exertion increases pain, fatigue and cognitive difficulties which characterize PEM. (1)
The interplay between reduced blood flow, lactic acid accumulation and autonomic dysfunction leads to a snowballing increase in symptoms and prolonged recovery times. Post-exertional malaise (PEM) has been compared to the metabolic changes that a marathon runner may experience following their race. ME/CFS patients have reduced reserves of Adenosine triphosphate (ATP) vital for mitochondrial energy production, and replenishment of ATP may take days.
PEM in ME/CFS involves complex metabolic changes including alterations in amino acid metabolism, with evidence of effects 24 hours after exercise and PEM activation. Non-essential amino acids, particularly those that can fuel the TCA cycle independently of pyruvate dehydrogenase (whose function is impaired in PEM) may become increasingly important for maintaining energy production during PEM episodes,(2)(3)(4) as shown by Fluge et al.(5)
The effect of catabolic metabolism from PEM, generally aligned with brainstem hypoperfusion, is associated with:
Muscle Protein Breakdown: PEM triggers muscle catabolism, releasing amino acids that are rapidly utilized, leading to systemic depletion.
Nitrogen Overload and Urea Cycle Stress, where increased utilization of aspartate depletes its systemic levels, impairing nitrogen clearance and contributing to metabolic stress.
Gut Barrier Dysfunction where appropriate: Increased intestinal permeability worsens systemic inflammation and impairs amino acid absorption.
Mitochondrial Dysfunction where reduced energy production forces reliance on amino acids for energy, further depleting their stores.
Neuroexcitation and BBB Breakdown: Elevated glutamate and impaired aspartate in the CNS, coupled with BBB dysfunction, drive excitotoxicity and localized hypermetabolism in the brain.
Brainstem Hypoperfusion
The brainstem, which consists of the midbrain, pons and medulla, has been implicated in ME/CIFS in many studies. It regulates the respiratory, cardiovascular, gastrointestinal, and neurological processes, which can be affected by long-COVID and similar disorders eg migraine and ME/CFS.
Griffith University showed in 2023 that the brainstem is larger in Long Covid and ME/CFS patients (8) and the brainstem demonstrates an imbalance of neurochemicals in this same group in 2024. (9) These changes correlate with the SPECT scan findings in ME/CFS, POTS and Long COVID found in our studies. The brainstem hypoperfusion is believed to be part of the same process of hypoperfusion and mitochondrial dysfunction that underpins the coat hanger pain of FMS, Long COVID, Fibromyalgia and POTS.
Figure 2. SPECT Scans showing brainstem hypoperfusion and cerebral hyperperfusion
These demonstrate the mixed hyperperfusion and brainstem hypoperfusion typical of POTS. Green represents normal perfusion. The blue areas reflect hypoperfusion, green normal, yellow, red then white increased metabolic activity/ hyperperfusion. The hyperperfusion is thought to be from endotheiliitis associated with intracranial vascular pressure as there is increasing evidence from our studies of impaired venous return causing a “backup” of venous pressure. Stagnant blood may play a part with its known activation of inflammatory cytokines causing the endotheleiitis. The scan image itself probably reflects increased levels of excitotoxic neurotransmitters affecting the brain through penetration through the dysfunctional BBB from the dysfunctional venous return.
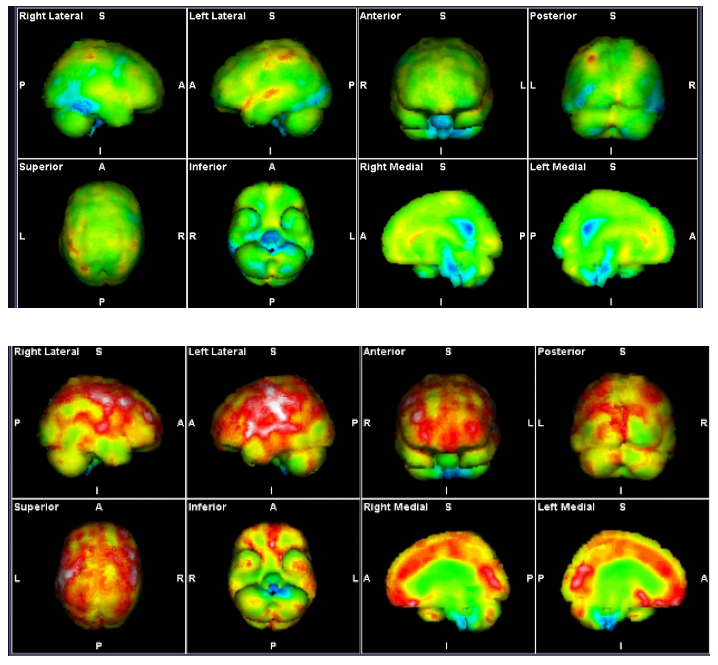
Source: Mermaid Molecular Imaging
In severe ME/CFS, Wirth et al (10) noted the reduction in blood flow in the brainstem from lying to sitting was 24.5%. They described the reduced blood flow to the brainstem causing neurological symptoms including impaired cognitive function or “brain fog.” This hypoperfusion can impact both the global and local regulation of blood flow in the brain. COVID-19 seriously affects the endothelium and there is evidence of chronic endothelial dysfunction in the post-Covid-syndrome similar to that in ME/CFS.(11)
Wirth et al (10) also reported muscle mitochondrial dysfunction, as evidenced by higher levels of pyruvate and lower levels of ATP and phosphocreatine in muscles, suggesting an impairment in muscle energy metabolism, which is also observed in ME/CFS, indicating a likely overlap in the pathophysiological mechanisms of these conditions.
Long Covid research by Appleman et al, (1) has also shown post-exertional malaise (PEM), with associated fatigue, pain and local and systemic metabolic disturbances, severe exercise-induced myopathy and tissue infiltration of amyloid-containing deposits in skeletal muscles of patients with long COVID.
Impaired Glymphatic Function
The impaired glymphatic function seen in Long COVID with its impairment of the paravascular space function also appears to have far-reaching effects on symptoms such as fatigue, brain fog and head pressure by CSF flow dysfunction. Glymphatic flow impairment exacerbates metabolic waste accumulation in the CNS, contributing to neuroinflammation and excitotoxicity. This further amplifies PEM symptoms, including cognitive dysfunction and fatigue.
Combining the research into the Glymphatic dysfunction, Intracranial Hypertension by Hulens (12), Bragee (13) and others, with brainstem hypoperfusion and the catabolic metabolism of coat hanger pain and PEM provides a satisfactory hypothesis for much of the symptomatology in all of these.
The use of brain SPECT scans in POTS, Long COVID and ME/CIFS has demonstrated brainstem hypoperfusion which may assist us in determining whether we may be dealing with mitochondrial dysfunction eg after Infectious Mononucleosis, or a vascular/autonomic dysfunction with brainstem hypoperfusion with the sequalae from this, or more commonly, a combination of both. - Brainstem Hypoperfusion, Coat Hanger pain and Post-Exertional Malaise in POTS and Long COVID. (14)
Hoel et al (6) described PEM in ME/CFS involving complex metabolic changes including alterations in amino acid metabolism, with breakdown (burn off) particularly of the branched amino acids leucine, isoleucine and valine, with evidence of affects 24 hours after exercise and PEM activation. Non-essential amino acids, particularly those that can fuel the TCA cycle independently of pyruvate dehydrogenase (whose function is impaired in PEM) may become increasingly important for maintaining energy production during PEM episodes.(15)(16)
This process generates a range of byproducts, some of which directly impact amino acid pathways and the urea cycle. These byproducts can accumulate, contributing to systemic metabolic dysfunction and exacerbating the symptoms of CFS and PEM.
Our findings are discussed in- Amino Acids, Essential Vitamin and Mineral Burn Off in Post Exertional Malaise (23)
Figure 3: Proposed mechanism of ME/CFS linked to amino acid catabolism.
Category I: amino acids are converted to pyruvate, and therefore depend on PDH to be further oxidized. These are alanine (Ala), cysteine (Cys), glycine (Gly), serine (Ser), and threonine (Thr).
Category II: amino acids that enter the oxidation pathway as acetyl-CoA, which directly and independently of PDH fuels the TCA cycle for degradation to CO2. These are isoleucine (Ile), leucine (Leu), lysine (Lys), phenylalanine (Phe), tryptophan (Trp), and tyrosine (Tyr).
Category III consists of amino acids that are converted to TCA cycle intermediates, thereby replenishing and supporting the metabolic capacity of the cycle- histidine (His), and proline (Pro)
The asterisks indicate the amino acids significantly reduced in ME/CFS patients.

Source: Fluge et al., Metabolic profiling indicates impaired pyruvate dehydrogenase function in myalgic encephalopathy/chronic fatigue syndrome. JCI Insight. 2016 Dec 22;1(21):e89376. doi: 10.1172/jci.insight.89376. PMID: 28018972; PMCID: PMC5161229.(5)
The combination of low GABA, low aspartic acid, elevated glutamate, ethanolamine, and lysine, alongside abnormal histidine, which I describe as the “Perfect Storm,” reflects a convergence of increased urea cycle utilization, muscle protein breakdown, mitochondrial dysfunction, and systemic inflammation. Similar amino acid abnormalities can be seen in a range of metabolic, inflammatory, and neurological conditions.
Mitochondrial Dysfunction with impairments in the TCA cycle and amino acid metabolism.
Neurotransmitter Imbalance, with excess excitatory signalling (glutamate) and reduced inhibitory signalling (GABA).
Metabolic Stress with dysregulation in nitrogen balance (aspartate, glutamate) and membrane metabolism (ethanolamine).
Ethanolamine is a component of cell membranes and involved in lipid metabolism. It is a precursor for phosphatidylethanolamine synthesis to phosphatidylcholine. Found very frequently in our amino acid studies, low urinary ethanolamine and high phosphoethanolamine levels suggest disruptions in phospholipid metabolism, potentially causing altered cell membrane composition, which could affect neuronal membrane stability and function, potentially impacting pain signalling.
Ethanolamine is involved in choline synthesis, which is important for acetylcholine production, a neurotransmitter involved in pain modulation. This dysfunction exacerbates the metabolic stress, neuroinflammation, and barrier breakdown observed in conditions like PEM, CFS, and brain hypoperfusion, and is associated with phospholipid dysfunction.
Lysine in preliminary studies is the most commonly effected amino acid. Lysine intake is crucial for maintaining healthy collagen function, which supports the structural integrity of skin, tendons, bones, and other connective tissues. Deficiencies can lead to weakened connective tissues, delayed wound healing, and other related health issues.
Other amino acids may be abnormal, such as proline. Proline is a major component of collagen, and high plasma levels may signal increased collagen turnover or connective tissue remodelling. High proline also contributes to glutamate accumulation, exacerbating excitotoxicity and limiting downstream utilization in the TCA cycle
The Urea Cycle
The urea cycle and the citric acid cycle are independent cycles but are linked. The urea cycle converts toxic nitrogenous compounds to excretable urea in five biochemical reactions. It is also the source for endogenous arginine, ornithine and citrulline production. The process mainly takes place in the liver, partly in the mitochondria and partly in the cytoplasm of the hepatocytes. The entire process converts two amino groups, one from NH+, and 1 from aspartate, and a carbon atom from HCO− to the relatively nontoxic excretion product urea. This occurs at the cost of four "high-energy"phosphate bonds (3 ATP hydrolyzed to 2 adenosine diphosphate (ADP) and one adenosine monophosphate (AMP). (46)
Figure 4: The Urea Cycle
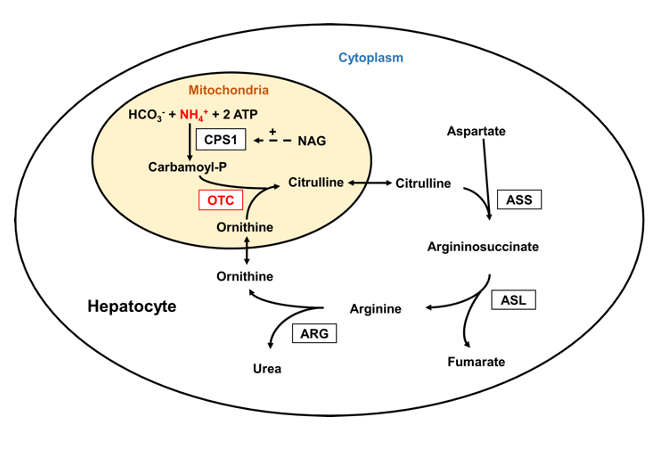
Source: Laemmle, Alexander; Gallagher, Renata C.; Keogh, Adrian; Stricker, Tamar; Gautschi, Matthias; Nuoffer, Jean-Marc; et al. (2016). Urea cycle.. PLOS ONE. Figure. https://doi.org/10.1371/journal.pone.0153358.g001 (47)
Nicotinamide and SIRT4
Sirtuin 4 (SIRT4) is a mitochondrial protein that inhibits mitochondrial glutamate dehydrogenase 1(GDH) activity, thereby downregulating insulin secretion in response to amino acids, as well as PDH activity. This regulation prevents overactivation of PDHC under nutrient surplus conditions, preserving mitochondrial homeostasis.
Nicotinamide, as a precursor for NAD⁺, directly influences SIRT4 activity and mitochondrial metabolism. The interplay between nicotinamide, NAD⁺, and SIRT4 can influence the observed amino acid imbalances. Nicotinamide, by restoring NAD⁺ levels and modulating SIRT4, could break this "perfect storm" by enhancing mitochondrial efficiency, normalizing amino acid metabolism, and balancing neurotransmitter levels. Nicotinamide is available in a number of forms, eg normal Vitamin B3, nicotinamide, has been used for skin cancer modulation for many years.
Restoring the NAD+ levels can enhance the proline oxidation pathway, helping to regulate the proline-glutamate interconversion. NAD+ also supports the malate-aspartate shuttle, helping to address aspartate depletion. NAD+ supports urea cycle efficiency, helping clear ammonia that may accumulate from amino acid imbalances (e.g., proline and lysine dysregulation).
Clinic studies have found introducing liposomal nicotinamide riboside has positive effects on fatigue and amino acid patterns, including stabilizing of ethanolamine function. Liposomal delivery systems significantly enhance the bioavailability and clinical efficacy of supplements, including nicotinamide riboside (NR) for mitochondrial and metabolic dysfunction (as seen in CFS) and vitamin B3 (nicotinamide/niacin) for skin health and cancer prevention.
Therapeutic Potential of Nicotinamide and SIRT4 Modulation
Restores NAD⁺ Levels by boosting NAD⁺ which supports mitochondrial metabolism, improving TCA cycle function, amino acid metabolism, and neurotransmitter balance.
Inhibits SIRT4 (Indirectly) at high concentrations, relieving GDH inhibition and enhancing glutamate clearance.
Supports Neurotransmitter Balance as improved glutamate metabolism increases GABA synthesis, addressing excitotoxicity and anxiety.
Supports urea cycle efficiency, potentially reducing brain fog.
Stabilizes Amino Acid Metabolism as replenished mitochondrial function improves aspartate production, lysine metabolism, and ethanolamine utilization.
Reduces Mast Cell Activation by reducing inflammatory signalling, stabilizing histidine/histamine balance.
GABA and low Aspartic acid
In managing CFS patients with abnormal amino acid profiles, the addition of nicotinamide riboside has shown promise in restoring levels of lysine, ethanolamine, and valine to normal ranges. However, the persistently low urinary aspartic acid levels, believed to be reflective of underlying hypoxia during post-exertional malaise (PEM), remain a challenge. This imbalance with glutamate can lead to neuroexcitation, and while lowering dietary glutamate intake can be beneficial, additional pathways and management strategies may be helpful.
To address low aspartic acid levels, Dunstan et al (17) proposed supplementation with N-acetylcysteine (NAC) may help increase aspartate levels by promoting the conversion of cysteine to cystine, which can then be used to synthesize aspartate. (18) The potential benefits of NAC for ME/CFS patients, particularly in addressing oxidative stress and potentially improving symptoms, are promising. However, the risks, while generally mild, should not be overlooked. (19)
Supporting the urea cycle with supplements like arginine or citrulline may help increase aspartate production, although the important research by Weigel et al (18) found that the role of nutritional intake and supplement use on ME/CFS patients’ health-related quality of life remains unclear, dietary changes and the use of supplements appear to be of value to ME/CFS patients.
With the addition of GABA to the amino acid assays, low levels of GABA are being consistently seen. There is evidence supports the idea that increasing GABA levels can help counteract the excitatory effects of glutamate and potentially reduce neuroexcitation. There may be insufficient conversion of glutamate to GABA due to impaired glutamate decarboxylase (GAD) activity or pyridoxal-5’-phosphate (active B6) deficiency. If B6 levels are low (and care must be taken to ensure avoiding neurotoxicity from excess B6, Pyridoxal-5’-phosphate (P5P) starting at 25 to 50 mg daily may be helpful.
Additionally, there is evidence for the role of magnesium in glutamate regulation. Magnesium plays a significant role in regulating glutamate activity, as magnesium acts as a natural NMDA receptor antagonist, which can help regulate glutamate activity, and by blocking the NMDA receptor channel, magnesium can reduce excessive glutamate signalling.
Magnesium's action at NMDA receptors can protect against glutamate-induced excitotoxicity, which is implicated in various neurological disorders. (20) Magnesium can influence the release of neurotransmitters, including glutamate, potentially helping to maintain proper excitatory/inhibitory balance.(20) The impact of magnesium supplementation may vary depending on an individual's baseline magnesium levels and overall neurological health. Suggested is magnesium glycinate form, 200 to 400mg daily. Maximum dose in women is 350mg.(44)
While the evidence supports the potential benefits of increasing GABA levels (and magnesium supplementation) in regulating glutamate activity, the relationship between these neurotransmitters is complex and context-dependent. While increasing GABA levels and magnesium supplementation show promise in regulating glutamate excitation, their effects can be nuanced and dependent on various factors. For example, in some cases, GABA can be excitatory, particularly during early development or in certain pathological conditions. (97) The effectiveness of GABA in counteracting glutamate excitation can depend on factors such as intracellular chloride concentrations and the expression of specific transporters. (21)(22)
CoQ10 at 100 to 300 mg daily has been shown may support mitochondrial function alongside nicotinic acid, the combination more effective than each alone in neuroprotection, blocking ATP deletions and lactate increases.(24) Yet nicotinamide and NAD+, nor randomly applied supplementation, while may improve fatigue, will not solve the metabolic problems.
Persistent aspartate depletion and glutamate dysregulation in PEM
Preliminary results in amino acid testing after initial treatment with liposomal nicotinamide to modulate the SIRT4 signalling. Persistent aspartate depletion shown in follow up testing, while still at a preliminary stage, points to a fundamental metabolic bottleneck in PEM, where aspartate is disproportionately consumed in an effort to maintain cellular energy homeostasis. This aligns with the broader metabolic crisis hypothesis of PEM, where a fragile energetic system is pushed beyond its threshold following exertion.
At the core of this dysfunction, aspartate’s depletion is driving an unresolved secondary glutamate imbalance, even when absolute glutamate levels appear normal. This suggests that the pathology is not simply a matter of excess glutamate production but rather a failure of glutamate utilization, recycling, and compartmentalization.
The likely linking is:
Mitochondrial Stress & TCA Cycle Over-reliance on Aspartate
Aspartate is critical for TCA cycle anaplerosis, and its persistent depletion suggests a constant state of mitochondrial distress.
The malate-aspartate shuttle (MAS) is not keeping up with demand, leading to intracellular deficits.
Energy failure leads to compensatory glutamate dysregulation, as glutamate itself is a key metabolic intermediary.
Dysregulated Astrocyte-Glutamate Interactions
Astrocytes rely on aspartate for normal glutamate uptake and conversion to glutamine.
With depleted aspartate, astrocytic glutamate transporters (EAAT1/EAAT2) may be inefficient, prolonging glutamate's synaptic effects.
This could explain why patients exhibit glutamate toxicity symptoms (brain fog, cognitive impairment, hypersensitivity) even with normal glutamate levels.
Impaired Urea Cycle Function Leading to Secondary Ammonia Stress
Aspartate is essential for nitrogen clearance through the aspartate-argininosuccinate pathway in the urea cycle.
Persistent aspartate depletion → inefficient ammonia detoxification → compensatory increase in glutamine synthesis
This could create a pseudo-normalization of glutamate levels while actually worsening the glutamate-glutamine cycle.
High glutamine → osmotic and neurotransmitter dysregulation, worsening PEM symptoms.
Excitotoxicity & Energy Crisis in the Context of PEM
Since aspartate contributes to both excitatory neurotransmission and mitochondrial energy production, its depletion may create a mismatch between neuronal energy availability and excitation.
This could explain the classic PEM delay effect—initial exertion isn’t immediately catastrophic, but over time, as aspartate reserves deplete, glutamate dysregulation and energy failure manifest with a worsening of symptoms.
It may explain why PEM can last for days/weeks, because replenishing aspartate stores in a dysfunctional system is inherently slow, leading to prolonged energy crisis states
Malate-Aspartate Shuttle.
Figure 5. Malate-Aspartate Shuttle

Source: Amadeo, Hanspers,K., Coort,S., Weitz. Malate-aspartate shuttle (WP4315), WikiPathways.(48)
Amadeo et al (48) describe “The malate-aspartate shuttle (sometimes also the malate shuttle) is a biochemical system for translocating electrons produced during glycolysis across the semipermeable inner membrane of the mitochondrion for oxidative phosphorylation in eukaryotes. These electrons enter the electron transport chain of the mitochondria via reduction equivalents to generate ATP. The shuttle system is required because the mitochondrial inner membrane is impermeable to NADH, the primary reducing equivalent of the electron transport chain. To circumvent this, malate carries the reducing equivalents across the membrane.”(48)
Aspartate and possible use in POTS, Long COVID and CFS
Increasing anecdotal reports have been reporting the use of a peptide containing L-Aspartate and other amino acids promoting the healing of different tissues, including skin, muscle, bone, ligament and tendon in many animal studies. Reports suggest there may be:
reduction in post exertional malaise (PEM)
vascular repair
improved gut mucosal repair and gut barrier integrity with reduced reactive dietary events,
accelerated muscle repair
enhanced epithelial regeneration
reduced oxidative stress
improved mitochondrial function
improved mental health
These reports are anecdotal and the product is considered “performance-enhancing” and not approved for use in Australia. Concern in its use revolve around lack of human clinical trials, unknown potential long-term effects and potential for misuse. The peptide also may not be ideal for all POTS, Long COVID, and CFS patients, particularly those with pre-existing excitotoxicity, dysautonomia, or vascular instability. (42)(43)
It's apparent ability to control the PEM symptoms highlights the importance of improving the Malate-Aspartate Shuttle function. Similarly the importance of improving collagen metabolism that is inherently associated with the amino acid dysregulation in CFS, POTS and Long COVID needs to be considered.
L-Aspartate is a major component of the peptide. L-Aspartate plays a critical role in fibroblast proliferation, extracellular matrix (ECM) synthesis, and cellular signalling. While it is unlikely all the benefits of the peptide can be attributed to L-Aspartate, other ingredients include proline and glycine may have a role in collagen synthesis and fibroblast activity, and lysine in extra-cellular matrix, exponents of its use in tissue repair focus on its holistic activity rather than the action of individual amino acids.
L-Aspartate is the biologically active form involved in TCA cycle metabolism, the malate-aspartate shuttle, glutamate balance, and neurotransmitter clearance. It is the most effective form for mitochondrial function, glutamate clearance, and metabolic regulation.
L-Aspartate is essential for gut repair and mitochondrial metabolism but could exacerbate excitotoxicity, dysbiosis, or serotonin-related gut issues in certain conditions such as Inflammatory bowel disease eg Crohn’s and Ulcerative Colitis, SIBO, Coeliac disease and possibly MCAS. It may be beneficial in leaky gut/gut barrier dysfunction, constipation-dominant IBS and where increased fibroblast activity is warranted.
When mitochondrial ATP production declines (e.g., in hypoxia, mitochondrial dysfunction, or PDH deficiency), Na⁺ gradients collapse, directly impairing glutamate and aspartate transport. PDH dysfunction causes pyruvate accumulation and increased lactate production, shifting the cell into a pseudo-hypoxic state. This triggers reactive oxygen species (ROS) and inflammation, which can downregulate transporter expression or impair function. Reduced ATP availability impairs preload regulation and vascular tone, while dysregulated glutamate/aspartate metabolism affects autonomic control.
L-Aspartate supplementation is thought to bypass TCA cycle defects via reductive carboxylation of glutamine.(39)(40) It is involved in collagen synthesis (proline and hydroxyproline precursor), and the Malate-Aspartate Shuttle regulates NADH transfer for ECM remodelling. Increased L-Aspartate availability correlates with higher fibroblast-derived collagen deposition, but excess may contribute to fibrosis, especially under hypoxic or inflammatory conditions.
Fibroblasts are the primary collagen-producing cells in connective tissue. Their activation is regulated by amino acid metabolism (Aspartate, Proline, Glutamine), growth factors (TGF-β, VEGF, IGF-1), oxidative stress, mitochondrial function and NMDA receptor-mediated calcium signalling
Aspartic acid influences fibroblast proliferation, differentiation, and ECM remodelling, but D-Aspartate (D-Asp) and L-Aspartate (L-Asp) affect fibroblast function differently.
Aspartate is a key amino acid involved in glutamate homeostasis (via aspartate-glutamate exchange), collagen production, the malate-aspartate shuttle (essential for mitochondrial NADH transfer), the urea cycle, and neurotransmitter balance (aspartate is both an excitatory neurotransmitter and a precursor for glutamate) (38) Disruptions in aspartate/glutamate ratios can result in excitotoxicity, mitochondrial inefficiency, and impaired ammonia detoxification—common features in POTS, Long COVID, chronic fatigue syndrome (CFS), fibromyalgia, and neurodegenerative diseases.
Aspartate can theoretically act as an excitotoxin and while aspartate is not as potent as glutamate in excitatory neurotransmission, high doses may still trigger neuroexcitability, making its use in patients with neuroinflammation and seizures speculative. Its potential effectiveness should be governed by the severity of mitochondrial impairment, urea cycle dysfunction and efficiency of glutamate clearance as well as comorbidities.
To consider its use, Aspartate needs to be combined with supportive metabolic interventions, and consideration of comorbidities in the choice of which form of Aspartate, D- or L-aspartate and what other metabolic changes may be required if this supplement may be appropriate. It should be considered as an adjunctive therapy rather than stand alone, ideally with a strong knowledge of each patient’s metabolic profiling.
D-Aspartate (D-Asp) is primarily found in neuroendocrine tissues, is involved in testosterone regulation, NMDA receptor activity, and neurodevelopment, but has limited metabolic roles in the TCA cycle. It is not efficiently utilized in energy metabolism (it does not enter the TCA cycle). Evidence suggests D-Asp may increase testosterone levels,(32) but it also interacts with the hypothalamic-pituitary-adrenal (HPA) axis.
Elevated NMDA activity is linked to cognitive dysfunction, fatigue, and neuroinflammation, which are core features of CFS. D-Aspartate may act as an NMDA receptor agonist, potentially exacerbating glutamate excitotoxicity in patients with dysautonomia, neuroinflammation, or neurodegenerative conditions (e.g., POTS, Long COVID, ME/CFS), which could worsen symptoms like brain fog, insomnia, sensory hypersensitivity, and autonomic dysfunction.
D-Aspartate is primarily a signalling molecule, influencing fibroblasts through NMDA receptor activation modulating calcium influx in fibroblasts, VEGF upregulation promoting angiogenesis and connect tissue remodelling, and neuroendocrine crosstalk altering fibroblast hormonal responses.
Excess D-Asp may however over-activate NMDA receptors in fibroblasts, potentially leading to fibrotic tissue remodelling, dysregulated ECM degradation, increased oxidative stress and mitochondrial dysfunction.
Testosterone plays a significant role in the HPA axis, particularly in response to stress where it regulates stress responses and maintains hormonal balance, blunting cortisol responses, but also increasing ACTH.(33) This interaction has distinct implications for POTS and Intracranial Hypertension (ICH). While potentially increasing ICH, it also may stabilize autonomic dysfunction in appropriate patients.(34)(35)
The association with testosterone brings another link of aspartate dysfunction into the complex issues in POTS. Low D-aspartate contributes to low testosterone and may increase (cause) autonomic dysfunction, with potential benefits of supplementation by improving vascular tone and autonomic regulation by restoring testosterone levels, NMDA overactivity and stabilization of noradrenaline release and reduced POTS-associated tachycardia. The evidence for this at present is unclear and the impact speculative.
POTS predominantly affects females, yet the underlying sex-based differences remain poorly understood. D-Aspartate acts as a positive regulator of testosterone synthesis in Leydig cells.(36) Men have higher levels of D-Aspartate, which plays a role in neuroendocrine regulation. This may confer protection against autonomic instability by supporting baroreceptor sensitivity, vascular tone, and cerebral perfusion.
D-Aspartate deficiency could alter NMDA receptor function and cerebrovascular control, making the brain more vulnerable to hypoperfusion and autonomic instability. This imbalance may increase susceptibility to POTS and other dysautonomias.
POTS, CFS and Long COVID patients have brainstem hypoperfusion. Inadequate perfusion of the brainstem and baroreceptor pathways could explain the exaggerated heart rate response and autonomic instability in POTS. Low D-Aspartate levels could disrupt NMDA-mediated neurovascular control, reducing blood flow to critical autonomic centres. NMDA receptor dysfunction leads to impaired CSF drainage and venous outflow, increasing intracranial pressure (ICP). High ICP further disrupts cerebral autoregulation, worsening POTS symptoms upon standing.
D-Aspartate has been shown to increase testosterone production, particularly in younger males with low baseline levels. However, in CFS patients, the risks of excitotoxicity, oxidative stress, and hormonal disruption probably outweigh the benefits. It does present an area for further investigation.
Patients with Chronic Fatigue Syndrome (CFS/ME) and low testosterone present unique metabolic challenges, particularly in mitochondrial function, neuroinflammation, and hormonal dysregulation. The choice of L-Aspartate vs. D-Aspartate is important as each form impacts different physiological pathways. While there is insufficient evidence to ensure the safety of D-Aspartate in CFS, it may be of potential use to support low testosterone, but probably only in a closely monitored environment.
Overall, L-Aspartate appears to be the preferred form in CFS, given its mitochondrial and metabolic benefits. While there are no accurate guidelines for the use of L-Aspartate, the limited available literature suggests:
L-Aspartate as Sodium L-Aspartate (to aid absorption and sodium-dependent transport) in CFS 2–4 g/day (divided into 2 doses, morning and evening). Start low (1g/day) and increase gradually to monitor tolerance, although there is limited data on dosage.
Co-factors for mitochondrial support, particularly magnesium glycinate form, (200 to 400mg) daily, may influence the release of neurotransmitters, including glutamate, potentially helping to maintain proper excitatory/inhibitory balance.(20) Maximum dose in women is 350mg.
Taurine may be of value (1-2 gm/day) to help stabilize glutamate and enhance autonomic balance,(45) although this study was limited by sample size.
Foods with high L-Aspartate and low D-Aspartate include soy protein, egg white, chicken breast, turkey, fish, beef, lentils, black beans, peanuts and almonds. Foods with high D-Aspartate include aged cheeses, fermented soy, and cured meats due to protein racemization
Nutraceutical approaches (e.g., NAD+ precursors, CoQ10) may enhance mitochondrial function synergistically.
Overall, these pathways are providing valuable information for ongoing research. At the present time, I consider targeted metabolic solutions rather than haphazard use of peptides, or even supplements with the possible exception of magnesium, and nicotinamide where appropriate, plus close attention to diet.
Future Directions
Mechanistic studies to investigate the malate-aspartate shuttle’s role in PEM pathophysiology.
Amino acid profiling, expanding metabolic studies to include GABA, glutamine, and proline dynamics.
Clinical trials to evaluate targeted interventions, including L-aspartate and lip[osomal nicotinamide riboside, in controlled settings.
Investigate the actions of GLP-1 in increasing insulin (and thereby stimulating hepatic IGF-1 production) alongside the modulatory effects of amino acid-based products on IGF-1 receptor expression, that offer a promising avenue to restore metabolic stability. This integrated approach could help improve recovery from post-exertional malaise, providing a more comprehensive strategy for managing the multifaceted challenges of POTS.
Conclusion
In the complex metabolic dysfunction seen in POTS, the interplay between aspartate dysregulation, cortisol dysfunction, and nocturnal hypoglycaemia (under investigation) creates a challenging metabolic environment that can compromise both energy production and neuroendocrine balance.
While detailed family history can provide some genetic risk information, individualized DNA through Dr Vittone provides far more information into the various gene mutations than the normal commercially available products, and have been providing valuable insights into these complex pathways. These can provide a springboard into nutraceutical treatments.
Understanding the metabolic, mitochondrial, and neuroimmune mechanisms underlying PEM provides a critical framework for developing personalized diagnostics and targeted therapies where Dr Vittone can provide valuable pathways to deal with the dysfunctional metabolic pathways. By focusing on amino acid metabolism, mitochondrial efficiency, and neurotransmitter balance, interventions such as NAD+ replenishment, SIRT4 modulation, and targeted amino acid support hold promise for mitigating PEM and improving outcomes in ME/CFS, POTS, and Long COVID.
By addressing aspartate metabolism, mitochondrial efficiency, and neurotransmitter imbalances, emerging therapies hold promise for improving outcomes in POTS, CFS, and related disorders. A personalized approach—grounded in metabolic profiling and evidence-based interventions—is essential for advancing the management of these complex conditions.
References:
1. Appelman, B., Charlton, B.T., Goulding, R.P. et al. Muscle abnormalities worsen after post-exertional malaise in long COVID. Nat Commun 15, 17 (2024). https://doi.org/10.1038/s41467-023-44432-3
2. McGregor NR, Armstrong CW, Lewis DP, Gooley PR. Post-Exertional Malaise Is Associated with Hypermetabolism, Hypoacetylation and Purine Metabolism Deregulation in ME/CFS Cases. Diagnostics (Basel). 2019 Jul 4;9(3):70. doi: 10.3390/diagnostics9030070. PMID: 31277442; PMCID: PMC6787670.
3. Germain A, Giloteaux L, Moore GE, Levine SM, Chia JK, Keller BA, Stevens J, Franconi CJ, Mao X, Shungu DC, Grimson A, Hanson MR. Plasma metabolomics reveals disrupted response and recovery following maximal exercise in myalgic encephalomyelitis/chronic fatigue syndrome. JCI Insight. 2022 May 9;7(9):e157621. doi: 10.1172/jci.insight.157621. PMID: 35358096; PMCID: PMC9090259.
4. Glass KA, Germain A, Huang YV, Hanson MR. Urine Metabolomics Exposes Anomalous Recovery after Maximal Exertion in Female ME/CFS Patients. Int J Mol Sci. 2023 Feb 12;24(4):3685. doi: 10.3390/ijms24043685. PMID: 36835097; PMCID: PMC9958671.
5. Fluge Ø, Mella O, Bruland O, Risa K, Dyrstad SE, Alme K, Rekeland IG, Sapkota D, Røsland GV, Fosså A, Ktoridou-Valen I, Lunde S, Sørland K, Lien K, Herder I, Thürmer H, Gotaas ME, Baranowska KA, Bohnen LM, Schäfer C, McCann A, Sommerfelt K, Helgeland L, Ueland PM, Dahl O, Tronstad KJ. Metabolic profiling indicates impaired pyruvate dehydrogenase function in myalgic encephalopathy/chronic fatigue syndrome. JCI Insight. 2016 Dec 22;1(21):e89376. doi: 10.1172/jci.insight.89376. PMID: 28018972; PMCID: PMC5161229.
6. Hoel F, Hoel A, Pettersen IK, Rekeland IG, Risa K, Alme K, Sørland K, Fosså A, Lien K, Herder I, Thürmer HL, Gotaas ME, Schäfer C, Berge RK, Sommerfelt K, Marti HP, Dahl O, Mella O, Fluge Ø, Tronstad KJ. A map of metabolic phenotypes in patients with myalgic encephalomyelitis/chronic fatigue syndrome. JCI Insight. 2021 Aug 23;6(16):e149217. doi: 10.1172/jci.insight.149217. PMID: 34423789; PMCID: PMC8409979.
7. Vittone, V., Exelby, G. DNA Mutations the Underpin POTS and Long COVID. 2024. https://www.mcmc-research.com/post/dna-mutations-that-underpin-pots-and-long-covid
8. Thapaliya K, Marshall-Gradisnik S, Barth M, Eaton-Fitch N, Barnden L. Brainstem volume changes in myalgic encephalomyelitis/chronic fatigue syndrome and long COVID patients. Front Neurosci. 2023 Mar 2;17:1125208. doi: 10.3389/fnins.2023.1125208. PMID: 36937672; PMCID: PMC10017877.
9. Thapaliya K, Marshall-Gradisnik S, Eaton-Fitch N, Eftekhari Z, Inderyas M, Barnden L. Imbalanced Brain Neurochemicals in Long COVID and ME/CFS: A Preliminary Study Using MRI. Am J Med. 2024 Apr 6:S0002-9343(24)00216-X. doi: 10.1016/j.amjmed.2024.04.007. Epub ahead of print. PMID: 38588934.
10. Wirth, K.J., Scheibenbogen, C. & Paul, F. An attempt to explain the neurological symptoms of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. J Transl Med 19, 471 (2021). https://doi.org/10.1186/s12967-021-03143-3
11. Verger, A., Kas, A., Dudouet, P. et al. Visual interpretation of brain hypometabolism related to neurological long COVID: a French multicentric experience. Eur J Nucl Med Mol Imaging 49, 3197–3202 (2022). https://doi.org/10.1007/s00259-022-05753-5
12. Hulens M, Dankaerts W, Rasschaert R, Bruyninckx F, De Mulder P, Bervoets C. The Link Between Empty Sella Syndrome, Fibromyalgia, and Chronic Fatigue Syndrome: The Role of Increased Cerebrospinal Fluid Pressure. J Pain Res. 2023;16:205-219https://doi.org/10.2147/JPR.S394321
13. Bragée B, Michos A, Drum B, Fahlgren M, Szulkin R, Bertilson BC. Signs of Intracranial Hypertension, Hypermobility, and Craniocervical Obstructions in Patients With Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Front Neurol. 2020 Aug 28;11:828. doi: 10.3389/fneur.2020.00828. PMID: 32982905; PMCID: PMC7485557.
14. Exelby,G. Brainstem Hypoperfusion, Coat Hanger pain and Post-Exertional Malaise in POTS and Long COVID. 2024. https://www.mcmc-research.com/post/brainstem-hypoperfusion-coat-hanger-pain-and-post-exertional-malaise-in-pots-and-long-covid
15. Dingledine R, McBain CJ. Glutamate and Aspartate Are the Major Excitatory Transmitters in the Brain. In: Siegel GJ, Agranoff BW, Albers RW, et al., editors. Basic Neurochemistry: Molecular, Cellular and Medical Aspects. 6th edition. Philadelphia: Lippincott-Raven; 1999. : https://www.ncbi.nlm.nih.gov/books/NBK28252/
16. van den Pol AN, Wuarin JP, Dudek FE. Glutamate, the dominant excitatory transmitter in neuroendocrine regulation. Science. 1990 Nov 30;250(4985):1276-8. doi: 10.1126/science.1978759. PMID: 1978759.
17. Dunstan RH, Sparkes DL, Roberts TK, Crompton MJ, Gottfries J, Dascombe BJ. Development of a complex amino acid supplement, Fatigue Reviva™, for oral ingestion: initial evaluations of product concept and impact on symptoms of sub-health in a group of males. Nutr J. 2013;12:115. Published 2013 Aug 8. doi:10.1186/1475-2891-12-115
18. Weigel B, Eaton-Fitch N, Passmore R, Cabanas H, Staines D, Marshall-Gradisnik S. A preliminary investigation of nutritional intake and supplement use in Australians with myalgic encephalomyelitis/chronic fatigue syndrome and the implications on health-related quality of life. Food Nutr Res. 2021;65:10.29219/fnr.v65.5730. Published 2021 Jun 7. doi:10.29219/fnr.v65.5730
19. Assessment of N-Acetylcysteine as Therapy for Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. National Institute of Neurological Disorders and Stroke. https://www.ninds.nih.gov/health-information/clinical-trials/assessment-n-acetylcysteine-therapy-myalgic-encephalomyelitischronic-fatigue-syndrome
20. Sears SM, Hewett SJ. Influence of glutamate and GABA transport on brain excitatory/inhibitory balance. Exp Biol Med (Maywood). 2021;246(9):1069-1083. doi:10.1177/1535370221989263
21. Kilb W. When Are Depolarizing GABAergic Responses Excitatory?. Front Mol Neurosci. 2021;14:747835. Published 2021 Nov 24. doi:10.3389/fnmol.2021.747835
22. de Leon AS, Tadi P. Biochemistry, Gamma Aminobutyric Acid. [Updated 2023 May 1]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK551683/
23. Exelby, G. Amino Acids, Essential Vitamin and Mineral Burn Off in Post Exertional Malaise. 2024. https://www.mcmc-research.com/post/amino-acid-essential-vitamin-and-mineral-burn-off-in-post-exertional-malaise
24. Beal MF, Henshaw DR, Jenkins BG, Rosen BR, Schulz JB. Coenzyme Q10 and nicotinamide block striatal lesions produced by the mitochondrial toxin malonate. Ann Neurol. 1994 Dec;36(6):882-8. doi: 10.1002/ana.410360613. PMID: 7998775.
25. Postural Orthostatic Tachycardia Syndrome. John Hopkins Medicine. https://www.hopkinsmedicine.org/health/conditions-and-diseases/postural-orthostatic-tachycardia-syndrome-pots
26. Zhao S, Tran VH. Postural Orthostatic Tachycardia Syndrome. [Updated 2023 Aug 7]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK541074/
27. Kizilbash SJ, Ahrens SP, Bruce BK, Chelimsky G, Driscoll SW, Harbeck-Weber C, Lloyd RM, Mack KJ, Nelson DE, Ninis N, Pianosi PT, Stewart JM, Weiss KE, Fischer PR. Adolescent fatigue, POTS, and recovery: a guide for clinicians. Curr Probl Pediatr Adolesc Health Care. 2014 May-Jun;44(5):108-33. doi: 10.1016/j.cppeds.2013.12.014. PMID: 24819031; PMCID: PMC5819886.
28. Kato K, Sullivan PF, Evengård B, Pedersen NL. Chronic Widespread Pain and Its Comorbidities: A Population-Based Study. Arch Intern Med. 2006;166(15):1649–1654. doi:10.1001/archinte.166.15.1649
29. Grach,S et al. Overlapping conditions in Long COVID at a multisite academic center. Frontiers in Neurology. 2024. doi: 10.3389/fneur.2024.1482917
30. Lindheimer JB, Stegner AJ, Wylie GR, et al. Post-exertional malaise in veterans with gulf war illness. Int J Psychophysiol. 2020;147:202-212. doi:10.1016/j.ijpsycho.2019.11.008
31. Baraniuk JN, Adewuyi O, Merck SJ, et al. A Chronic Fatigue Syndrome (CFS) severity score based on case designation criteria. Am J Transl Res. 2013;5(1):53-68.
32.Roshanzamir F, Safavi SM. The putative effects of D-Aspartic acid on blood testosterone levels: A systematic review. Int J Reprod Biomed. 2017 Jan;15(1):1-10. PMID: 28280794; PMCID: PMC5340133.
33.Rubinow, D., Roca, C., Schmidt, P. et al. Testosterone Suppression of CRH-Stimulated Cortisol in Men. Neuropsychopharmacol 30, 1906–1912 (2005). https://doi.org/10.1038/sj.npp.1300742
34. O'Reilly MW, Westgate CS, Hornby C, et al. A unique androgen excess signature in idiopathic intracranial hypertension is linked to cerebrospinal fluid dynamics. JCI Insight. 2019;4(6):e125348. Published 2019 Mar 21. doi:10.1172/jci.insight.125348
35. Boris JR, McClain ZBR, Bernadzikowski T. Clinical Course of Transgender Adolescents with Complicated Postural Orthostatic Tachycardia Syndrome Undergoing Hormonal Therapy in Gender Transition: A Case Series. Transgend Health. 2019;4(1):331-334. Published 2019 Nov 20. doi:10.1089/trgh.2019.0041
36.Nagata Y, Homma H, Lee JA, Imai K. D-Aspartate stimulation of testosterone synthesis in rat Leydig cells. FEBS Lett. 1999 Feb 12;444(2-3):160-4. doi: 10.1016/s0014-5793(99)00045-9. PMID: 10050750.
37. Chang CH, Tsai WC, Hsu YH, Pang JH. Pentadecapeptide BPC 157 enhances the growth hormone receptor expression in tendon fibroblasts. Molecules. 2014;19(11):19066-19077. Published 2014 Nov 19. doi:10.3390/molecules191119066
38. Holeček M. Aspartic Acid in Health and Disease. Nutrients. 2023;15(18):4023. Published 2023 Sep 17. doi:10.3390/nu15184023
39. Cheng, CT., Qi, Y., Wang, YC. et al. Arginine starvation kills tumor cells through aspartate exhaustion and mitochondrial dysfunction. Commun Biol 1, 178 (2018). https://doi.org/10.1038/s42003-018-0178-4
40. Hart ML, Quon E, Vigil ABG, Engstrom IA, Newsom OJ, Davidsen K, Hoellerbauer P, Carlisle SM, Sullivan LB. Mitochondrial redox adaptations enable alternative aspartate synthesis in SDH-deficient cells. Elife. 2023 Mar 8;12:e78654. doi: 10.7554/eLife.78654. PMID: 36883551; PMCID: PMC10027318.
41. Seeley, MC., O’Brien, H., Wilson, G. et al. Novel brain SPECT imaging unravels abnormal cerebral perfusion in patients with postural orthostatic tachycardia syndrome and cognitive dysfunction. Sci Rep 15, 3487 (2025). https://doi.org/10.1038/s41598-025-87748-4
42. Deek SA. BPC 157 as Potential Treatment for COVID-19. Med Hypotheses. Published online November 9, 2021. doi:10.1016/j.mehy.2021.110736
43. Seiwerth S, Brcic L, Vuletic LB, Kolenc D, Aralica G, Misic M, Zenko A, Drmic D, Rucman R, Sikiric P. BPC 157 and blood vessels. Curr Pharm Des. 2014;20(7):1121-5. doi: 10.2174/13816128113199990421. PMID: 23782145.
44. Berkheiser, K. How much Magnesium should you take per day? Healthline. 2023. https://www.healthline.com/nutrition/magnesium-dosage
45. McLeay Y, Stannard S, Barnes M. The Effect of Taurine on the Recovery from Eccentric Exercise-Induced Muscle Damage in Males. Antioxidants (Basel). 2017;6(4):79. Published 2017 Oct 17. doi:10.3390/antiox6040079
46. Urea Cycle. Wikipedia. https://en.wikipedia.org/wiki/Urea_cycle
47. Laemmle, Alexander; Gallagher, Renata C.; Keogh, Adrian; Stricker, Tamar; Gautschi, Matthias; Nuoffer, Jean-Marc; et al. (2016). Urea cycle.. PLOS ONE. Figure. https://doi.org/10.1371/journal.pone.0153358.g001
48. Amadeo, Hanspers,K., Coort,S., Weitz. Malate-aspartate shuttle (WP4315), WikiPathways. https://www.wikipathways.org/pathways/WP4315.html
Comments